RAD-tag mappingとは
RADはRestriction site Associated DNAを意味し、制限酵素が認識するサイトの近傍にある塩基配列を指します。ゲノム全体を解析対象とするのではなく、特定の制限酵素サイトの近傍のみを解析の対象とすることで、シークエンスをするコストやデータの処理に必要なコンピュータの能力を減らすことができます。
RAD-tagライブラリの作成手順
- ゲノムDNAを二種類の制限酵素(6塩基認識)で消化する。制限酵素は突出末端を形成するものを選ぶ。
- それぞれの制限酵素の突出末端に合うアダプターを結合させる。アダプターにはその後のシークエンスに必要な配列が含まれている。
- DNA断片のサイズ分画を行う。シーケンサーに適した長さのものにする。
- PCRで増幅する。DNA断片のうち両端に同じアダプターが結合しているもの(同じ制限酵素の認識配列があるもの)はPCRの増幅効率が悪いのでここで割合が大きく減少する。
Rによる数理解析
RAD-tag ライブラリをどのぐらいシークエンスするとどのぐらいの情報が得られるのかをRを使って計算してみました。
制限酵素処理により得られるDNA断片の長さの分布
制限酵素は6塩基を認識するものを二種類混ぜて使うとします。ゲノムの塩基配列を完全にランダムとすると任意の6塩基が出現する確率は1/46です。二種類の制限酵素のいずれかが認識する配列が出現する確率は2/46となります。
二つの制限酵素サイトの間の長さはr=1の負の二項分布に従うと考えられます。負の二項分布は「統計的に独立なペルヌーイ試行を行ったとき、r回の成功を得るのに必要な試行回数の分布」(Wikipediaより引用)と定義されています。
得られるDNA断片の長さを横軸、その出現割合を縦軸にとると次のようになります。
> p <- 2/4^6 > curve(dnbinom(x, size=1, prob=p), from=0, to=1e4, xlab='Length of DNA fragment', ylab='probability', main='Distribution of length of DNA fragments' )
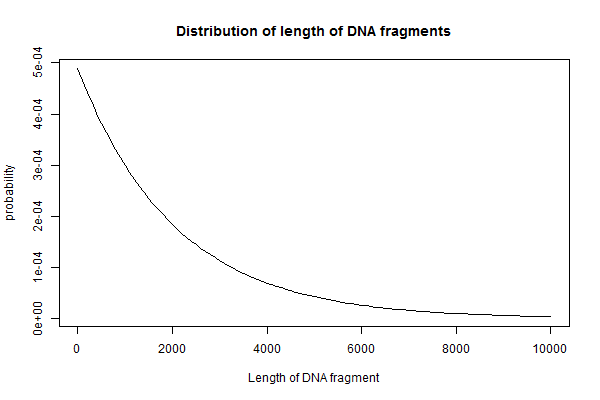
直感的には確率の逆数(2048)の長さがもっとも多くなるように思ってしまうかもしれませんが、実はそうはなりません。(ただし、実際の制限酵素は隣り合う配列は切断できないことが多いので10bpよりも短い断片ができることはないと思われます。)
DNA断片の選択
次に制限酵素処理で得られたDNA断片から1.両端が異なる制限酵素サイトであり、2.長さが300-500であるものを抽出します。1の条件はPCRで増幅されるものに該当し、2の条件は高速シーケンサーでシークエンスできるものに該当します。
Rでは次のように計算します。
> library_length <- 300:500 > pnbinom(max(library_length), size=1, prob=p) - pnbinom(min(library_length), size=1, prob=p) [1] 0.08033832
上記の計算により300-500bpの長さのDNA断片は約8%含まれていることになり、その内両端が異なる制限酵素サイトであるものは半分の約4%であると考えられます。
選択した断片を全部つなぐとどのぐらいの長さになるのでしょうか。断片の合計は「確率(存在割合) x 断片の長さ」の総和になります。そしてこれは期待値でもあります。負の二項分布の期待値は r / p で求められ、今回の場合はr=1ですから確率の逆数で2048となります。
300-500bpの範囲で確率と長さの積の和は以下のようにして求めることができます。
> library_density <- dnbinom(library_length, size=1, prob=p) > sum(library_length * library_density) [1] 32.17125 > 32.17125 * p [1] 0.01570862
二種類の制限酵素で処理したDNA断片のうち300-500bpのものを集めると全体の1.57%を集めてくることになります。
高速シーケンサーによる解析
ゲノムDNAを二種類の制限酵素で処理して得られる断片のうち両端が異なる制限酵素により切断されたもので、長さが300-500bpのものを集めてライブラリとしたとします。これをNGSでシークエンスして染色体マーカーを見つけたり、連鎖解析をするのに必要なシークエンス量を計算してみます。
次世代型シーケンサー(Illumina Genome Analyzerなど)はライブラリの片側を一定の長さシークエンスすることができます。これによりシークエンスできる領域はゲノム全体のどのぐらいになるのかを次のように計算します。
> sequence_cycle <- 50 > sum(sequence_cycle * library_density) * p [1] 0.001971681
この結果からゲノムの約0.2%の領域がシークエンスできることがわかります。ゲノムのサイズが400Mbの生物であれば約800kbが解析対象となります。多型が1000bpに1つである場合、800個のSNPが期待できると考えられます。
次に必要なリード数を計算してみます。ゲノムサイズを400Mbとすると二種類の制限酵素で切断したときの平均の長さが2048であることから2e5(20万)個の断片が生じていると考えられます。ライブラリに含まれるのはこのうち約4%ですから約7850種類のタグが期待できます。
> genome_size <- 400e6 > tags <- genome_size * p * (pnbinom(max(library_length), size=1, prob=p) - pnbinom(min(library_length), size=1, prob=p)) / 2 > tags [1] 7845.54
7850のタグのうちどれが出現するかはランダムであるので、n回出現するのに必要なリード数も負の二項分布に従うと考えられます。95%の断片がn回以上出現するために必要なリード数はqnbinom(0.05, size=n, prob=1/tags)となり、このnとリード数の関係をグラフにすると以下のようになります。
> curve(qnbinom(0.95, size=x, prob=1/tags), from=1, to=21, n=21, ylim=c(0, 2.5e5), xlab='n', ylab='reads', main='Required reads for n appearances at 5%' )
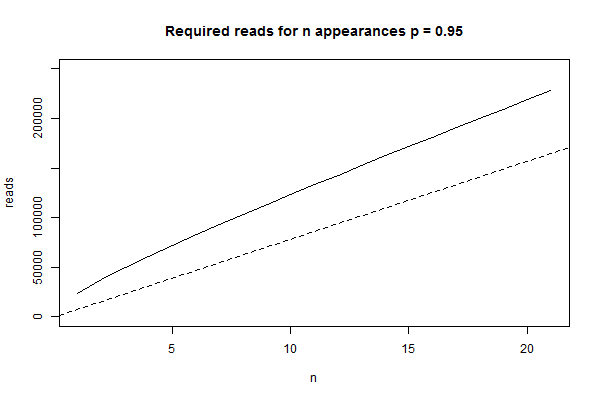
このグラフから95%のタグについて5回以上読もうと思うと約7.2万のリード数が必要であることがわかります。図中の点線はリード数から計算されるカバー数の期待値を示しており、期待値から推定するリード数の2倍程度が必要となることがわかります。
分離比の計算
あるタグの上にあるSNPはAとTの二つのパターンがあるとし、これをヘテロで持っていることがわかっているサンプルをn回シークエンスしたとします。このときにTが一回も出現しない確率は以下のように計算できます。
> curve(dbinom(0, x, p=0.5), from=0, to=10, n=11, xlab='coverage', ylab='probability', main='Probability of missing SNP' )
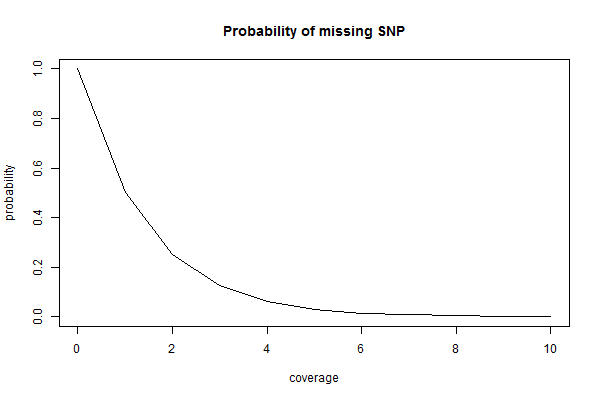
これは本来AとTがそれぞれ50%であるにも関わらず、Aが100%であると間違えて判定してしまう確率を示します。99%の確率で間違えないためには7回はシークエンスする必要がありそうです。
次に二種類の塩基が検出されたときにそれが誤りではないことを判断するために必要なシークエンス量について考えます。あるSNPにはAとTのパターンがあり、サンプルによりAのみ、Tのみ、AとTが50%ずつの場合があるとします。このサンプルをn回シークエンスしたときに、AまたはTがそれぞれ何回出現したらSNPのパターンを判定できるのでしょうか。下の図ではシーケンサーのエラー率を5%として、計算してみました。
cover <- 0:50
plot(cover, ylim=c(0,1), type='n', xlab='coverage', ylab='A/T ratio', main='Required coverage for SNP typing')
prob <- c(0.5, 0.95, 0.05)
color <- c('#00FF00FF', '#FF000080', '#0000FF80')
for(n in 1:length(prob)){
p <- prob[n]
polygon(
c(cover, rev(cover)),
c(
qbinom(0.95, size=cover, prob=p) / cover,
rev(qbinom(0.05, size=cover, prob=p) / cover)
),
col=color[n], border=NA
)
}
abline(v = 11, lty=2)
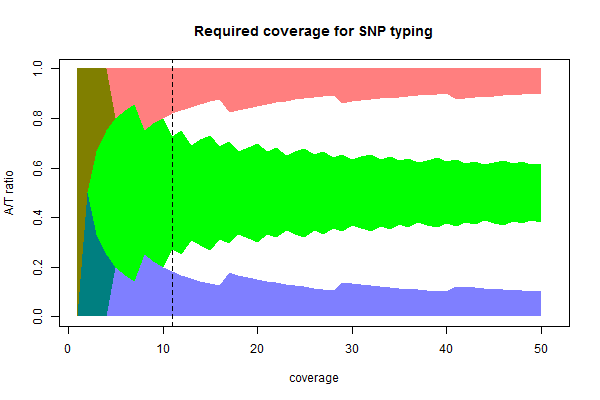
緑の領域はAとTが50%ずつであったときにAまたはTの出現回数のぶれる幅を示しています(90%信頼区間)。シークエンス回数(coverage)が増えるにつれ、ブレ幅が小さくなっていくことがわかります。20回シークエンスしたときには6-14回がA(またはT)であることが期待され、この範囲よりもAの回数が小さかったり大きかったりするときには50%であるという仮定を捨てたほうがよいことを示しています。
一方赤色の領域はどちらかの塩基が95%であると仮定してもおかしくない(90%の確率で起きうる)範囲を示しています。シークエンス回数が5回のとき4回Aが出現したとしてもAの存在割合を95%以上であると考えてよいことがわかります。
緑と赤(または青)が重なっているnの範囲ではSNPの判定を誤る可能性があります。明確に区別できるのはn=11(図中の点線)以上と考えられます。シークエンスの精度が99%程度であるならn=7以上でSNPを判定できるようになります。
まとめ
- シーケンサーの精度を95%とするとホモとヘテロを区別するためにはカバー数を11以上としなければならない。
- 95%のSNPについてカバー率を11以上とするためには、ライブラリに含まれるDNA断片の数の22倍程度のリード数が必要となる。
- シークエンス対象となるDNA断片の数は ゲノムサイズ x (2/46) x 0.04 と見積もることができる。
- 上記をまとめるとゲノムサイズ(Mb)の433倍のリード数があれば十分な解析ができると考えられます。
シーケンサーの精度を99%とすれば、276倍のリード数でよさそうです。
ライブラリの大きさ(多様性)をどのように担保するのかは重要そうです。同じ配列を繰り返し読んだときに、それらが最初のDNAのうち異なるものに由来するのかどうかはどうやったらわかるのかな。